


广东科学院微生物所吴清平院士团队:基于代谢组学和深度学习的单增李斯特菌快速鉴定新方法
发布时间:2023-06-16 浏览次数:1118 分享:
2022年4月26日,广东省科学院微生物研究所吴清平院士团队与暨南大学食品科学与工程系丁郁教授合作,提出了一种基于代谢组学和深度学习的单增李斯特菌快速鉴定新方法。相关研究成果以题为“Novel method for rapid identification of Listeria monocytogenes based on metabolomics and deep learning”发表在食品领域国际期刊《Food Control》(中科院1区Top期刊,IF=6.652)上。

成果简介
单核细胞增生李斯特菌是一种重要的食源性病原体,可导致免疫功能低下的个体、孕妇、胎儿、新生儿和老年人爆发李斯特菌病。李斯特菌病的死亡率为 20-30%,单核细胞增生李斯特菌是与食物中毒相关的第三大致死病因。因此,快速识别和检测李斯特菌对食品安全至关重要。基于质谱法的代谢组学可以作为检测病原体和腐败微生物的平台。然而,基于质谱的低分子量生物标志物的准确定量通常受到同位素标记标准和复杂规程的限制,不利于大规模应用。
本文开发了一种将代谢组学与深度学习相结合的新方法来鉴定单核细胞增生李斯特菌。建立了单核细胞增生李斯特菌三种潜在生物标志物的卷积神经网络(CNN)模型,预测准确率为82.2%。此外,使用伪靶向代谢组学方法获得了由29种代谢产物组成的代谢指纹,在层次聚类分析中可以成功区分六种常见李斯特菌。本研究中建立的CNN模型的二元分类器和多分类器,用于识别单核细胞增生李斯特菌和常见病原体,预测准确率分别为96.7%和96.3%。
实验流程
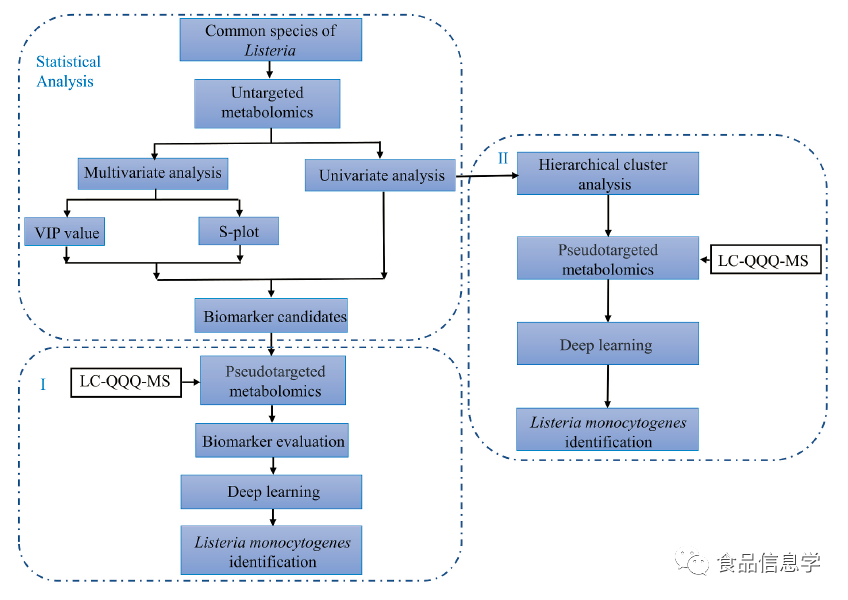
图1. 本研究方法工作流程图。
研究亮点
● 结合代谢组学和深度学习的平台用于病原体鉴定。
● 筛选了三个代谢潜在生物标志物用于单核细胞增生李斯特菌的鉴定。
● 建立了基于代谢指纹的CNN模型。
● 与传统的 LC-MS 方法相比,检测时间缩短至 12 分钟。
图文赏析
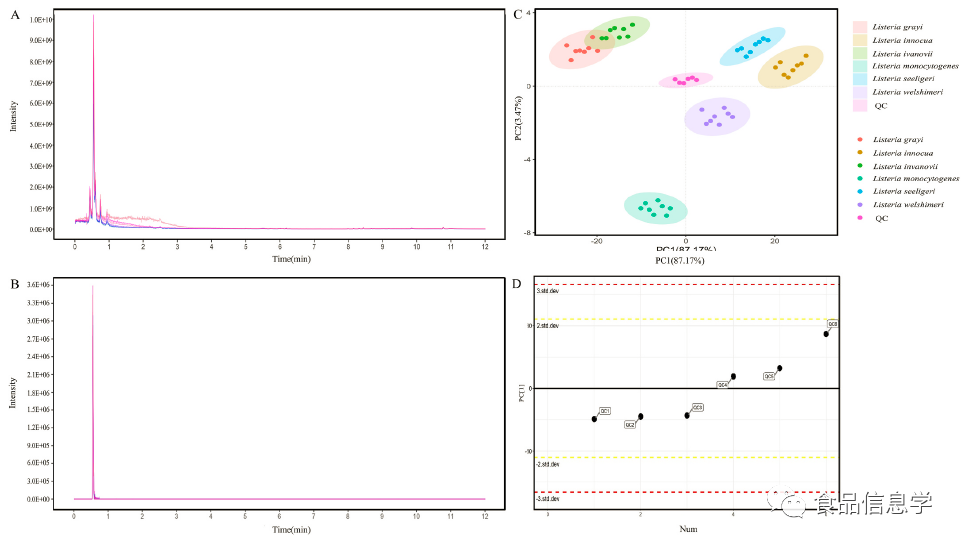
图2. 过程质量控制。(A) 正离子模式下质量控制(QC)样品的TIC图;(B) 正离子模式下质控样品的内标响应;(C) 六种常见李斯特菌的主成分分析;(D) QC样品的标准偏差。
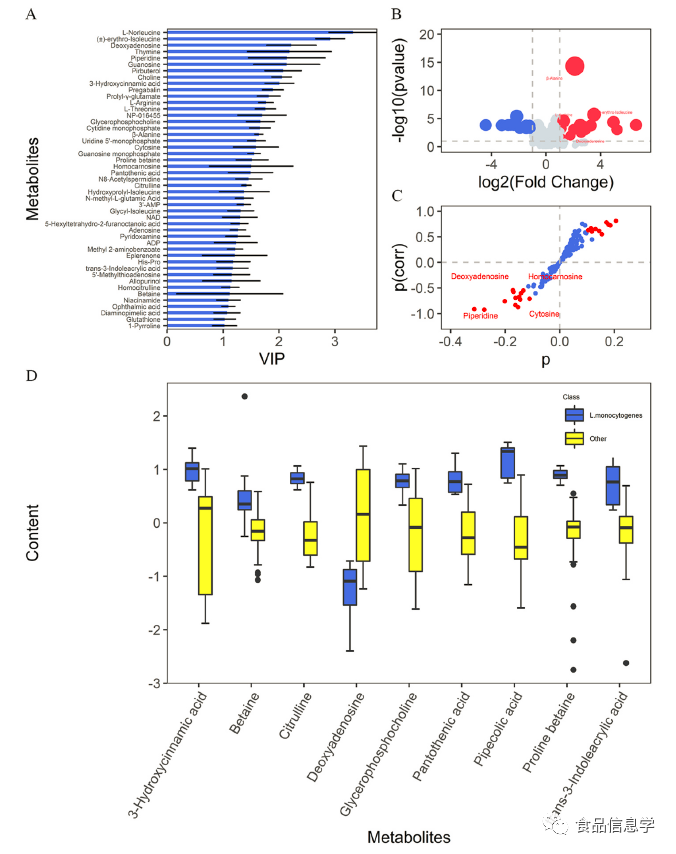
图3. 单变量和多变量分析。(A) VIP值的柱状图;(B) 单核细胞增生李斯特菌与其他菌株的火山图,其中红色和蓝色的点符合P≤ 0.05和log2(FC)>1;(C)具有显著代谢物的OPLS-DA模型的S图,其|p(corr)|>0.5和|p|>0.1;(D)候选生物标记物相关含量的箱线图。
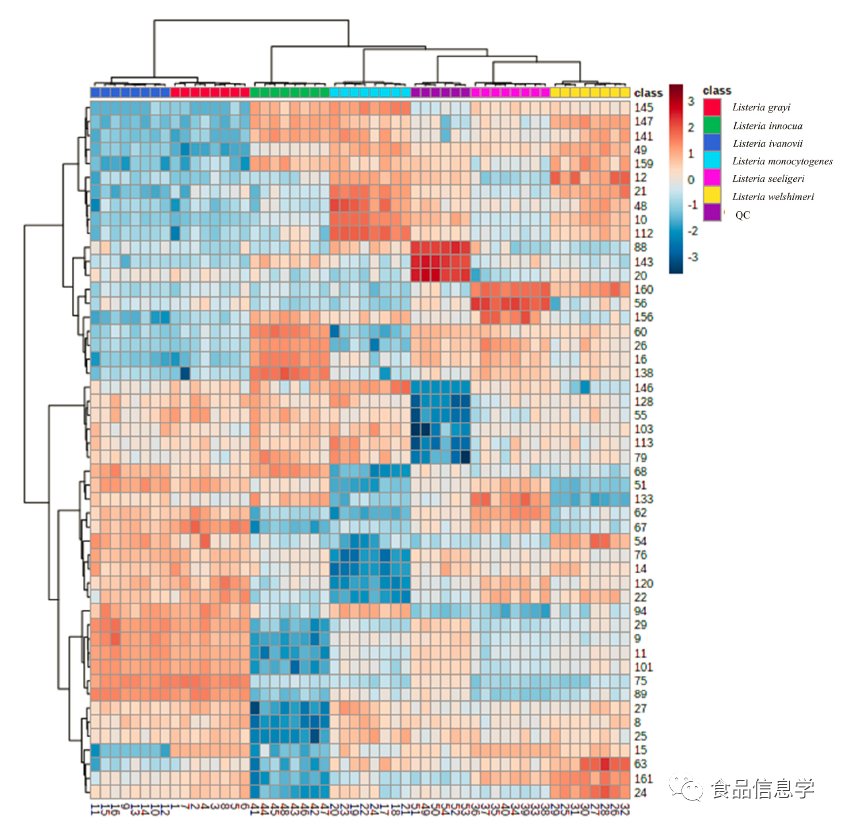
图4. 通过t检验生成的前50个代谢物的层次聚类热图。图中的每个彩色单元格对应于数据表中的浓度,行表示样本,列表式化合物。右侧的代谢物编号与表S2中代谢物的编号相对应。
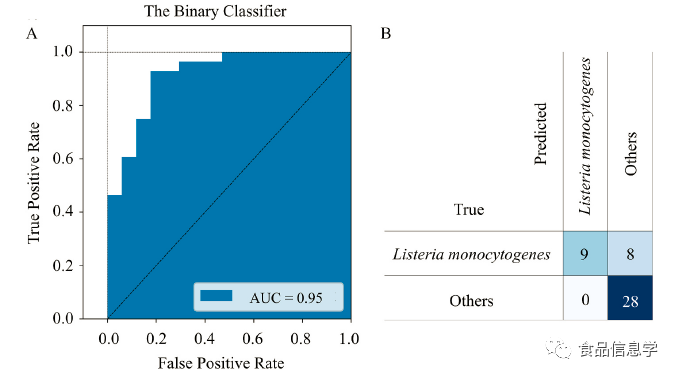
图5. 基于三种潜在生物标志物的卷积神经网络(CNN)模型的混淆矩阵图和接收器工作特性(ROC)曲线。(A) 测试集的ROC曲线(曲线下面积(AUC)为0.92);(B) 以混淆矩阵表示的测试集的预测结果;预测准确率为82.2%。
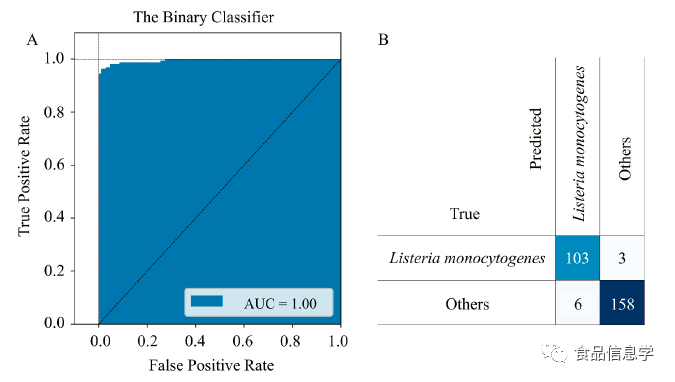
图6. 基于29个代谢特征作为代谢指纹的卷积神经网络(CNN)模型的混淆矩阵图和接收器工作特性(ROC)曲线。(A) 测试集的ROC曲线(曲线下面积(AUC)为0.98);(B)以混淆矩阵表示的测试集的预测结果。
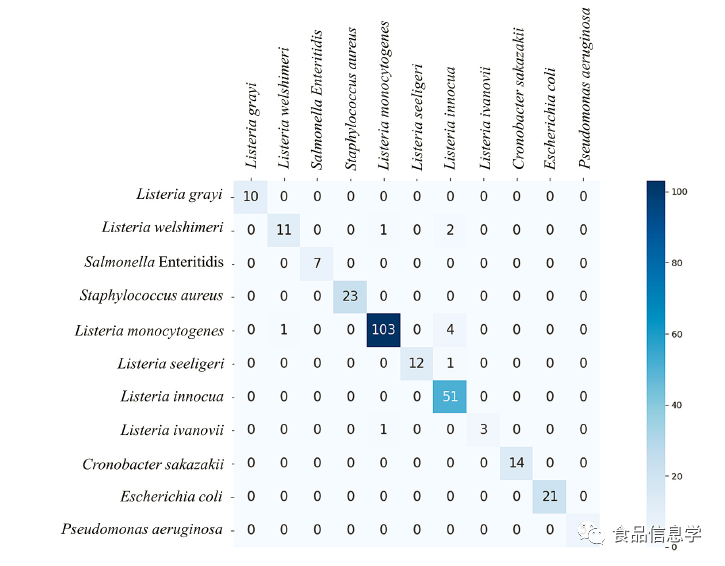
图7. 常见病原体测试集(30%样本)的卷积神经网络分类混淆矩阵图。
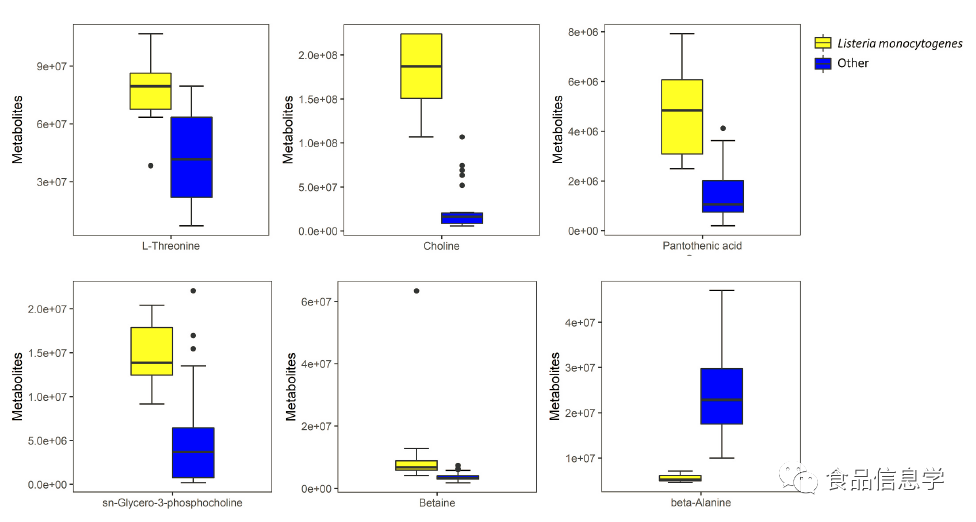
图8. 参与前四条通路的代谢物丰度变化。
表1. 内标响应差异。
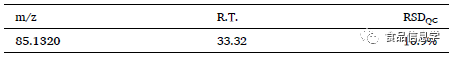
表2. 单增李斯特菌候选生物标志物的单变量和多变量分析结果。

表3. 经过LC-QQQ-MS确证后的单增李斯特菌候选生物标记物的单变量分析结果。

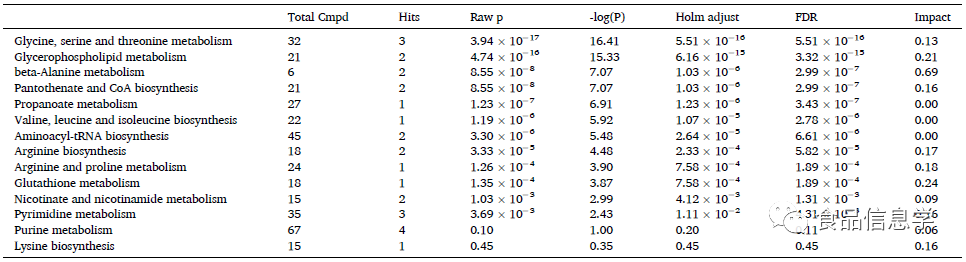
表4. 通路分析结果。
研究结论
本研究开发了一种新的病原体识别方法,包括使用基于特征代谢物LC-QQQ-MS指纹的深度学习模型,并使用伪靶向代谢组学进行潜在生物标记物验证。该策略是代谢组学领域的一个新发展。筛选了9种差异代谢物作为候选生物标记物,并使用LC-QQQ-MS验证了3种代谢物是可以在物种水平上识别单核细胞增生李斯特菌的潜在生物标记物。基于三种潜在生物标志物的CNN模型的最高预测准确率为82.2%。然后选取29个代谢特征作为代谢指纹,并将其与深度学习识别技术相结合,预测准确率提高到96.7%。此外,作者利用CNN模型对常见病原菌进行了鉴定,预测准确率为96.3%。
原文链接:https://doi.org/10.1016/j.foodcont.2022.109042

